这篇文章主要讲解了“如何使用ChIPpeakAnno进行peak注释”,文中的讲解内容简单清晰,易于学习与理解,下面请大家跟着小编的思路慢慢深入,一起来研究和学习“如何使用ChIPpeakAnno进行peak注释”吧!
ChIPpeakAnno是一个bioconductor上的R包,针对peak calling之后的下游分析,提供了以下多种功能
查找与peak区域最相邻的基因, 也支持自定义查找的特征,可以是exon,miRNA等
peak相邻基因的GO富集分析
提取peak及其周围区域的序列
在ChIPpeakAnno中,无论是peak区间信息还是基因组的注释信息,都通过toGRanges方法转化为R语言中的GRanges对象,以peak为例,bed格式的内容如下
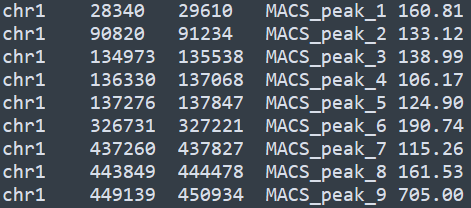
通过如下代码可以导入该信息
library(ChIPpeakAnno)
bed <- "peaks.bed"
gr <- toGRanges(bed, format="BED", header=FALSE)除了BED格式外,该方法也支持导入GTF格式的信息,只需要修改format参数即可。导入peak信息和基因组注释信息后就可以进行后续分析了。
当导入了多个样本的peak信息时,可以进行venn分析,用法如下
# 导入A样本的peak
bedA <- "sampleA_peaks.bed"
sampleA <- toGRanges(bedA, format="BED", header=FALSE)
# 导入B样本的peak
bedB <- "sampleB_peaks.bed"
sampleB <- toGRanges(bedB, format="BED", header=FALSE)
# 求交集
ol <- findOverlapsOfPeaks(sampleA, sampleB)
# 绘制venn图
makeVennDiagram(ol)结果示意如下
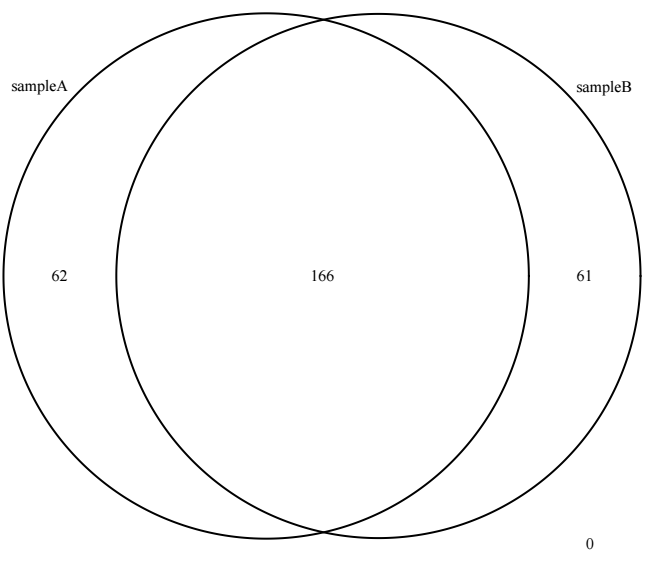
在进行venn分析时,会发现venn图上的个数加起来并不是输入的peak区间的总数,在默认
用法如下
library(BSgenome.Hsapiens.UCSC.hg19)
seq <- getAllPeakSequence(sampleA, upstream=20, downstream=20, genome=Hsapiens)
write2FASTA(seq, "sampleA.peaks.fa")提取到peak序列之后,可以进行motif分析,用法如下
# 用1号染色体的碱基分布当做背景
freqs <- oligoFrequency(Hsapiens$chr1, MarkovOrder=3)
# oligoLength规定了motif的长度
os <- oligoSummary(seq, oligoLength=6, MarkovOrder=3,
quickMotif=TRUE, freqs=freqs)
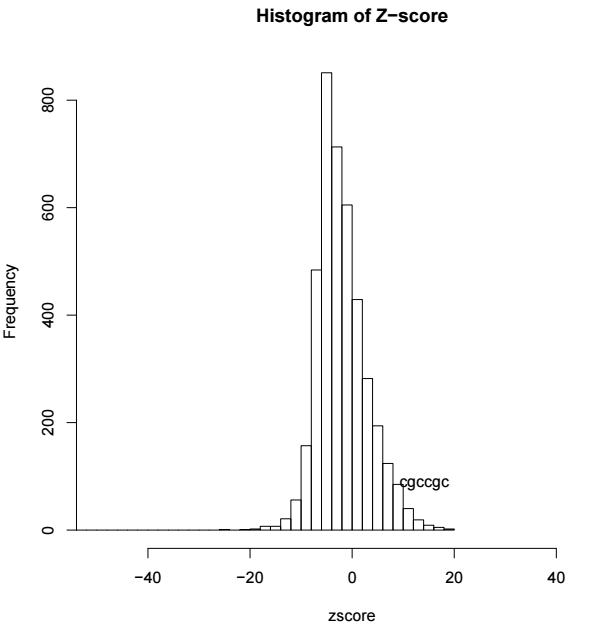
zscore <- sort(os$zscore)
# 绘制所有6个碱基组合的频率分布图
h <- hist(zscore, breaks=100, xlim=c(-50, 50), main="Histogram of Z-score")
# 频率最大的碱基组合即为motif的结果
text(zscore[length(zscore)], max(h$counts)/10,
labels=names(zscore[length(zscore)]), adj=1)结果示意如下
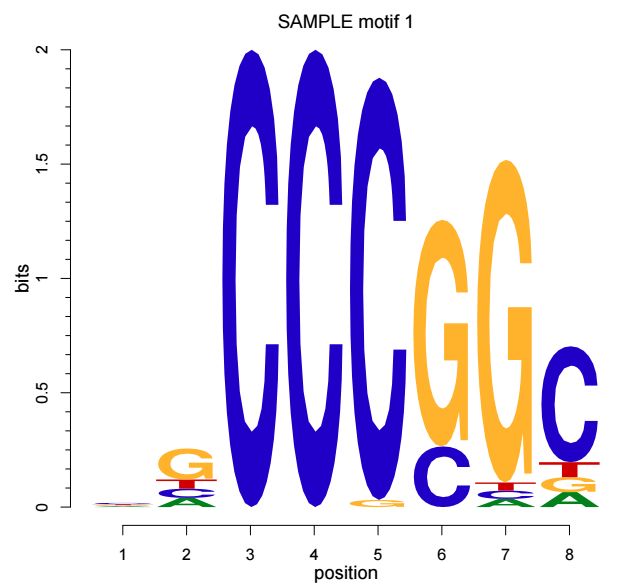
还可以通过motifStack这个R包绘制motif的sequence logo, 用法如下
library(motifStack)
pfms <- mapply(function(.ele, id)
new("pfm", mat=.ele, name=paste("SAMPLE motif", id)),
os$motifs, 1:length(os$motifs))
motifStack(pfms[[1]])输出结果示意如下
首先是peak在基因组各个特征区间的分布比例,用法如下
library(TxDb.Hsapiens.UCSC.hg19.knownGene)
aCR<-assignChromosomeRegion(sampleA, nucleotideLevel=FALSE,
precedence=c("Promoters", "immediateDownstream",
"fiveUTRs", "threeUTRs",
"Exons", "Introns"),
TxDb=TxDb.Hsapiens.UCSC.hg19.knownGene)
barplot(aCR$percentage, las=3)输出结果如下所示
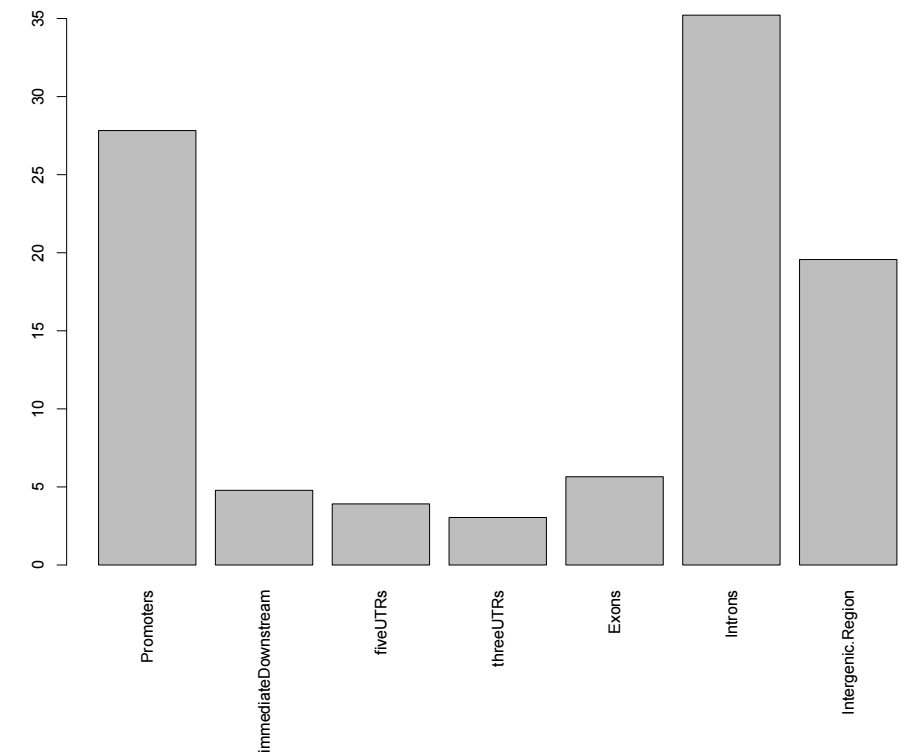
然后进行peak关联基因的注释,用法如下
# 准备基因组注释信息
library(EnsDb.Hsapiens.v75)
annoData <- toGRanges(EnsDb.Hsapiens.v75, feature="gene")
# 进行
overlaps.anno <- annotatePeakInBatch(sampleA,
AnnotationData=annoData,
output="nearestLocation"
)
library(org.Hs.eg.db)
overlaps.anno <- addGeneIDs(overlaps.anno,
"org.Hs.eg.db",
IDs2Add = "entrez_id")
pie1(table(overlaps.anno$insideFeature))输出结果示意如下
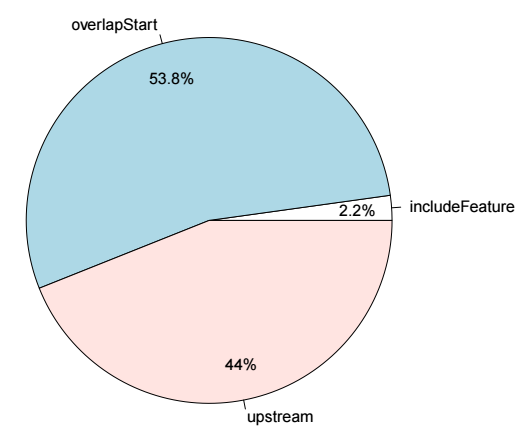
在使用annotatePeakInBatch进行注释时,默认查找距离peak最近的基因,也可以修改output的值,overlapping代表与peak区域存在overlap的基因,设置成这个值之后就会将与peak区间存在overlap的基因作为关联基因了,此外还有多种取值,适用不同条件,具体可以参考函数的帮助文档。
进行完基因注释之,得到peak关联的基因,就可以进行后续的功能富集分析,用法如下
over <- getEnrichedGO(overlaps.anno, orgAnn="org.Hs.eg.db",
maxP=.05, minGOterm=10,
multiAdjMethod="BH", condense=TRUE)ChIPpeakAnno提供了一条完整的peak下游分析功能,包括基因注释,富集分析,motif分析等等,是一个非常强大的工具,以上只是基本用法,更多用法和细节请参考官方文档。
感谢各位的阅读,以上就是“如何使用ChIPpeakAnno进行peak注释”的内容了,经过本文的学习后,相信大家对如何使用ChIPpeakAnno进行peak注释这一问题有了更深刻的体会,具体使用情况还需要大家实践验证。这里是亿速云,小编将为大家推送更多相关知识点的文章,欢迎关注!
免责声明:本站发布的内容(图片、视频和文字)以原创、转载和分享为主,文章观点不代表本网站立场,如果涉及侵权请联系站长邮箱:is@yisu.com进行举报,并提供相关证据,一经查实,将立刻删除涉嫌侵权内容。





