这期内容当中小编将会给大家带来有关怎么使用MutationalPatterns进行肿瘤突变频谱分析,文章内容丰富且以专业的角度为大家分析和叙述,阅读完这篇文章希望大家可以有所收获。
MutationalPatterns是一个bioconductor上的R包,可以用于肿瘤突变频谱的分析。肿瘤突变频谱针对点突变进行定义,A,T,C,G四种碱基两两突变,共有4X3=12种排列,考虑到正负链碱基配对原则,正链上的A->C突变,对应负链上为T->G, 所以进一步转换成了一个组合的问题,所以某个位点的突变可以划分为以下6种模式
C>A, 表示C>A和G>T两种
C>G, 表示C>G和G>C两种
C>T, 表示C>T和G>A两种
T>A,表示T>A和A>T两种
T>C,表示T>C和A>G两种
T>G,表示T>G和A>C两种
进一步考虑突变位点所处的序列上下文环境,即上下游各取一个碱基再加上突变位点的碱基,组成了3个碱基的motif, 可以有4X4X6=96种模式,每种模式的频率分布就是突变频谱。突变频谱可以当做一个肿瘤样本的特征,进行样本间的比较。通过MutationalPatterns包,可以方便的根据样本对应的VCF文件,提取突变频谱的信息,首先读取文件,代码如下
# 加载R包
> library(MutationalPatterns)
# 列出vcf的路径
> vcf_files <- c("sample1.vcf", "sample2.vcf")
# 设置vcf文件对应的样本名称
> sample_names <- c("sample1", "sample2")
# 加载参考基因组
> library(BSgenome.Hsapiens.UCSC.hg19)
> ref_genome <- "BSgenome.Hsapiens.UCSC.hg19"
# 读取vcf文件
> vcfs <- read_vcfs_as_granges(vcf_files, sample_names, ref_genome)读取完成之后,可以先统计下6种不同的点突变模式的分布,代码如下
> type_occurrences <- mut_type_occurrences(vcfs, ref_genome)
> plot_spectrum(type_occurrences)可视化结果示意如下
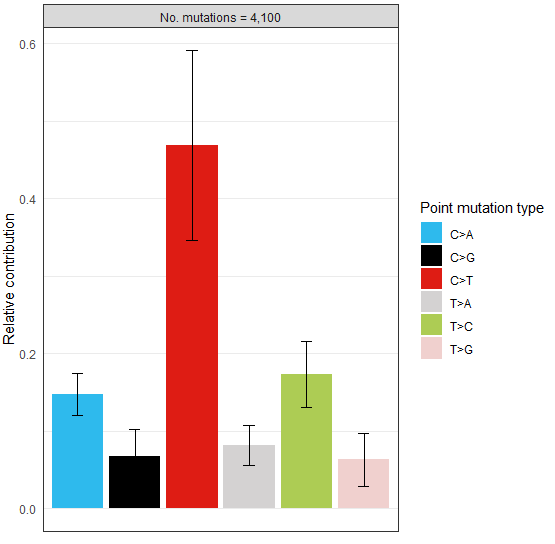
该R包经典的使用场景如下
根据vcf文件,计算每个样本中96种motif的频数,并可视化,代码如下
> mut_mat <- mut_matrix(vcf_list = vcfs, ref_genome = ref_genome)
> plot_96_profile(mut_mat[,c(1,2)], condensed = TRUE)可视化结果示意如下
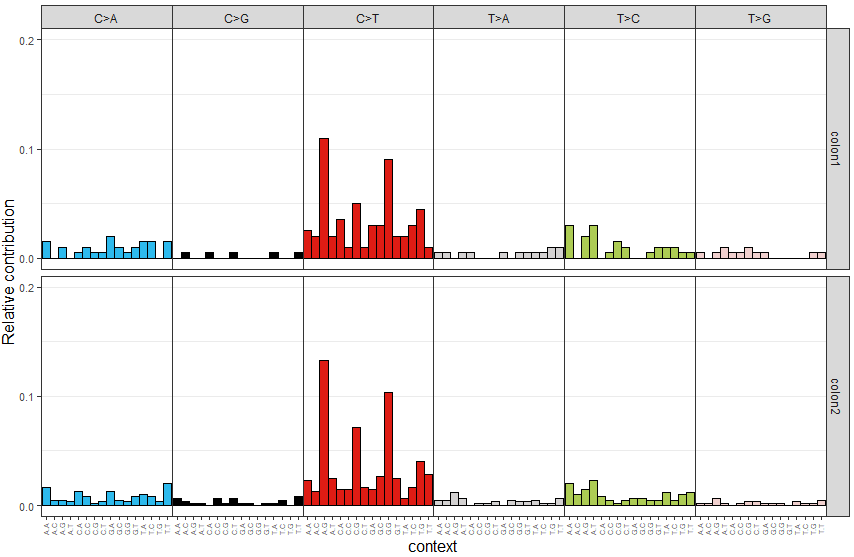
代码如下
> plot_compare_profiles(mut_mat[,1], mut_mat[,2], condensed = TRUE)
可视化结果示意如下
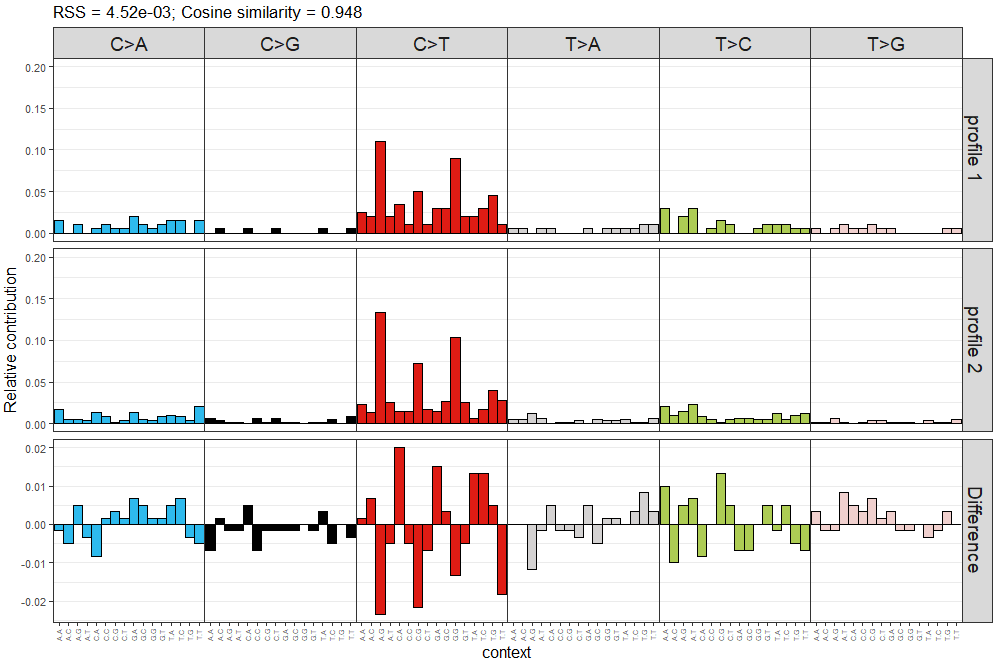
左上角的给出了两个频谱间cosine similarity相似度,图片中前两层分别对应两个需要比较的频谱,第三层为两个频谱的差异,直接用频率相减。
通过非负矩阵分解NMF算法,从原始的突变频谱中提取特征,称之为突变特征mutation signature,代码如下
> library(NMF)
> estimate <- nmf(mut_mat, rank=2:5, method="brunet", nrun=10, seed=123456)
> nmf_res <- extract_signatures(mut_mat, rank = 2, nrun = 10)
> colnames(nmf_res$signatures) <- c("Signature A", "Signature B")
> rownames(nmf_res$contribution) <- c("Signature A", "Signature B")
> plot_96_profile(nmf_res$signatures, condensed = TRUE)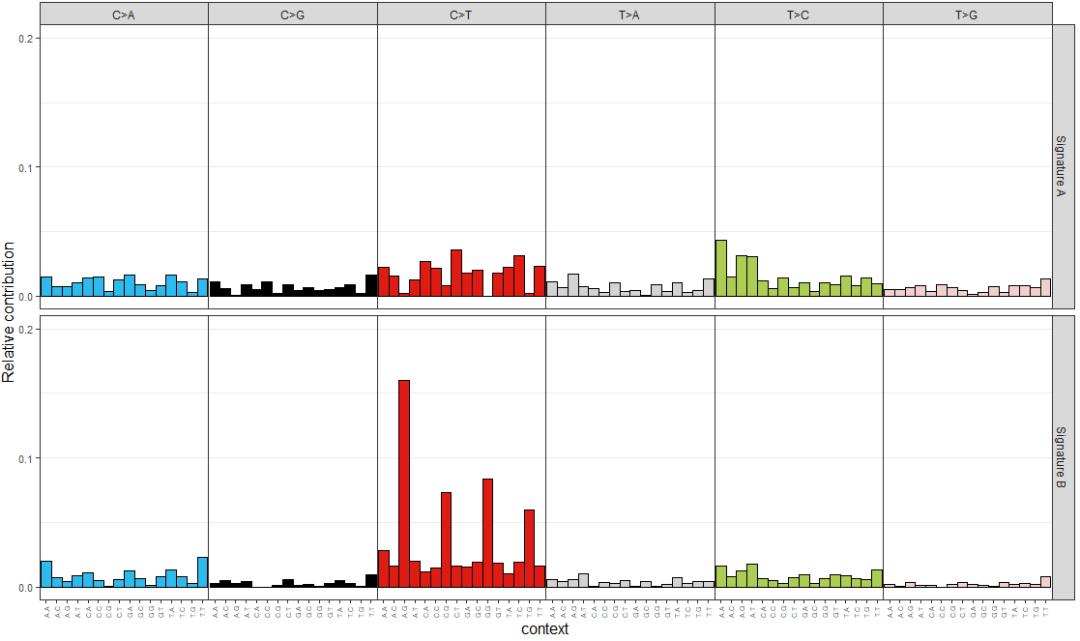
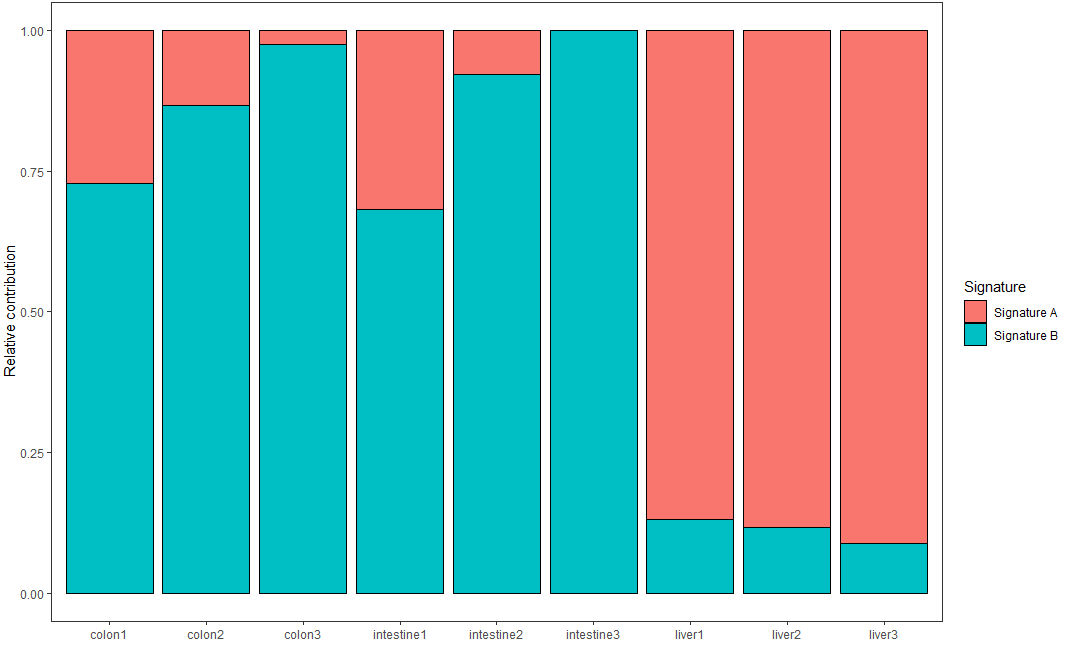
每个样本的突变频谱是不同突变特征组成的结果,通过如下代码可视化每个样本中不同突变特征的贡献率
plot_contribution(nmf_res$contribution, nmf_res$signature, mode = "relative")
可视化结果示意如下
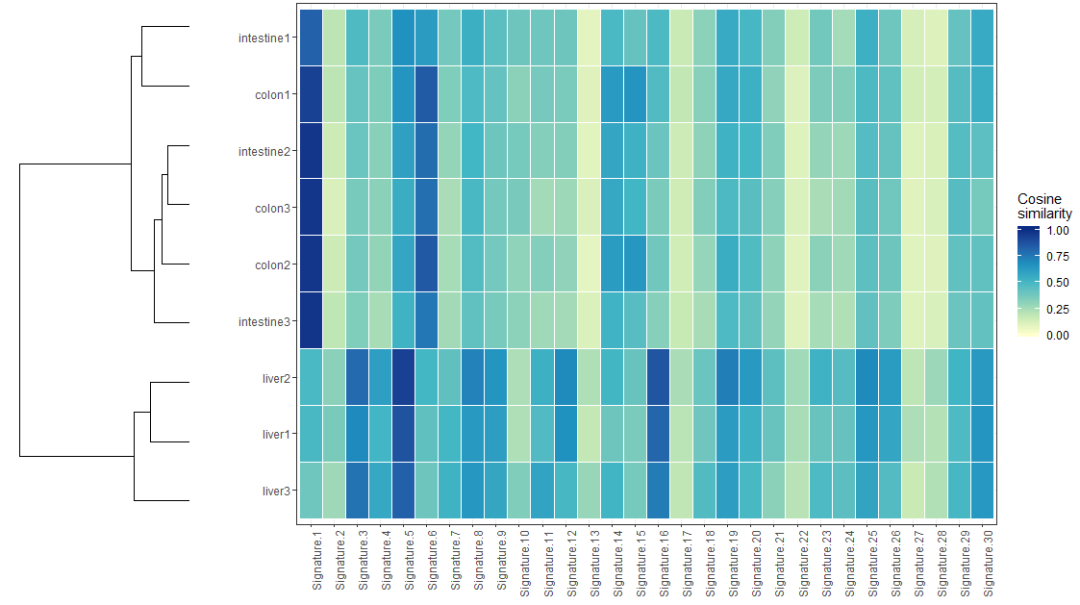
计算图片频谱间的cosine similarity相似度,结果用热图展现,代码如下
> cos_sim_samples_signatures = cos_sim_matrix(mut_mat, mut_mat)
> plot_cosine_heatmap(cos_sim_samples_signatures)可视化的结果示意如下
通过这个R包,可以轻松实现突变频谱的常见分析内容。
上述就是小编为大家分享的怎么使用MutationalPatterns进行肿瘤突变频谱分析了,如果刚好有类似的疑惑,不妨参照上述分析进行理解。如果想知道更多相关知识,欢迎关注亿速云行业资讯频道。
免责声明:本站发布的内容(图片、视频和文字)以原创、转载和分享为主,文章观点不代表本网站立场,如果涉及侵权请联系站长邮箱:is@yisu.com进行举报,并提供相关证据,一经查实,将立刻删除涉嫌侵权内容。





