这篇文章主要讲解了“如何使用ChIPseeker进行peak注释”,文中的讲解内容简单清晰,易于学习与理解,下面请大家跟着小编的思路慢慢深入,一起来研究和学习“如何使用ChIPseeker进行peak注释”吧!
ChIPseeker是使用的最广泛的peak注释软件之一,提供了以下多种功能
peak在染色体和TSS位点附近分布情况可视化
peak关联基因注释以及在基因组各种元件上的分布
获取GEO数据库中peak的bed文件
多个peak文件的比较和overlap分析
首先我们需要输入peak文件,支持两种格式,第一种是BED格式,最少只需要3列内容记录peak的染色体位置就可以了,示意如下

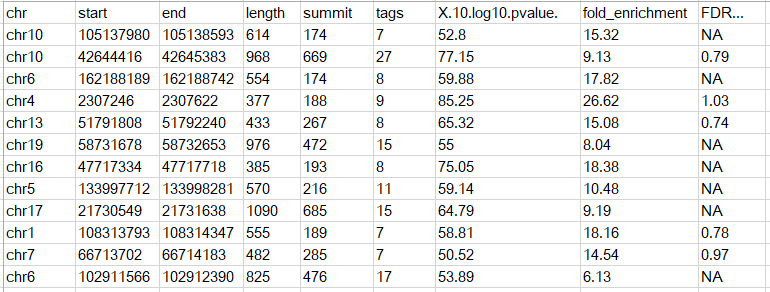
当然也可以有多余的列,只需要符合BED格式的标准即可;另外一种和MACS的peak calling输出结果类似,第一行为表头,示意如下
通过函数readPeaks读取peak文件,用法如下
peak <- readPeakFile("peak.bed")函数根据文件名称的后缀来判断是否为bed格式,建议BED格式的输入文件后缀统一成.bed, 当然压缩文件也是支持的,比如.bed.gz;如果不是BED格式的输入,文件名称则不能使用BED格式对应的后缀。
下面来详细看下几个主要功能的代码和结果展示
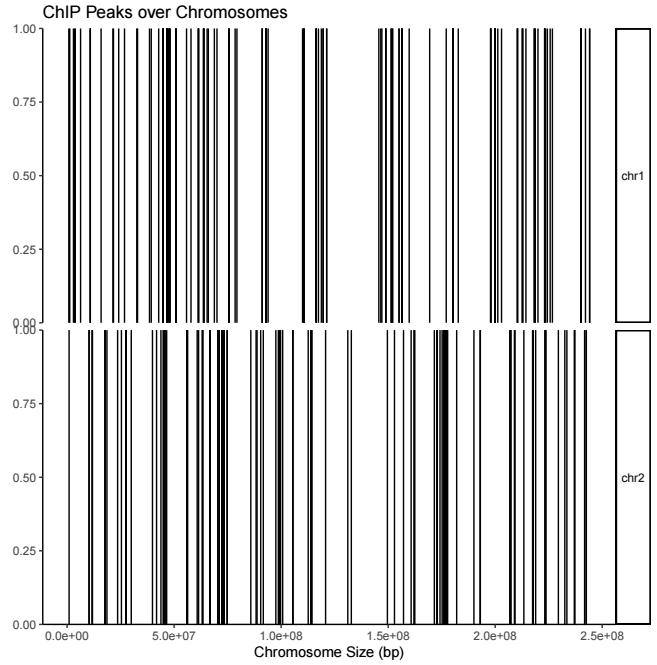
用法如下
covplot(peak, chr = c("chr1", "chr2"))输出结果示意如下
用法如下
library(TxDb.Hsapiens.UCSC.hg19.knownGene)
txdb <- TxDb.Hsapiens.UCSC.hg19.knownGene
# 定义TSS上下游的距离
promoter <- getPromoters(TxDb=txdb, upstream=3000, downstream=3000)
tagMatrix <- getTagMatrix(peak, windows=promoter)
tagHeatmap(tagMatrix, xlim=c(-3000, 3000), color="red")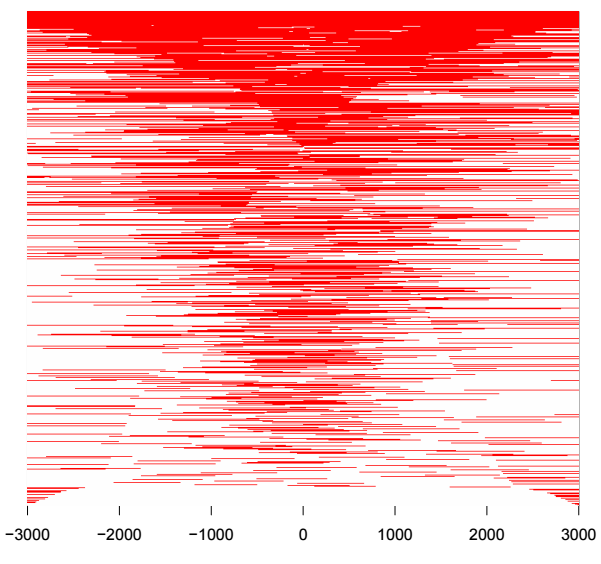
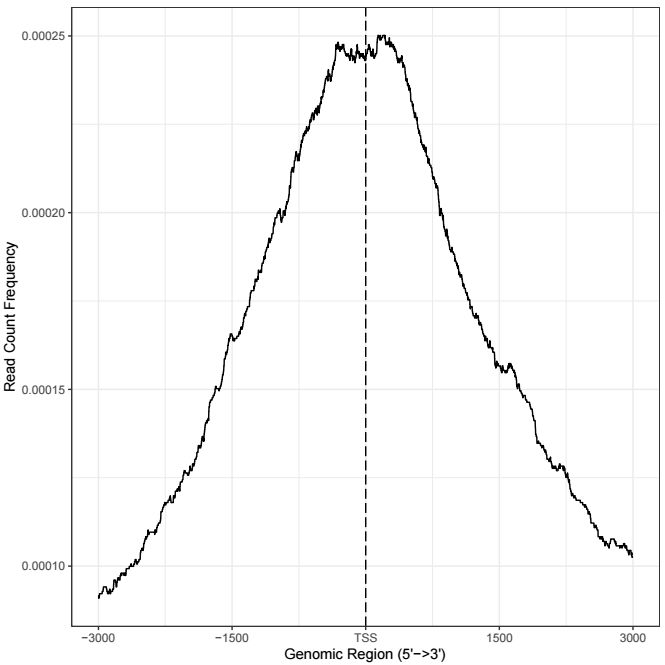
热图每一行代表一个基因,展示的是所有基因TSS两侧的分布,除了热图外,还可以对所有基因取均值,用折线图来展示TSS两侧分布情况,用法如下
plotAvgProf(
tagMatrix,
xlim=c(-3000, 3000),
xlab="Genomic Region (5'->3')",
ylab = "Read Count Frequency")输出结果示意如下
用法如下
peakAnno <- annotatePeak(
peak,
tssRegion = c(-3000, 3000),
TxDb = txdb,
annoDb = "org.Hs.eg.db")
write.table(
as.data.frame(peakAnno),
"peak.annotation.tsv",
sep="\t",
row.names = F,
quote = F)注释文件内容如下
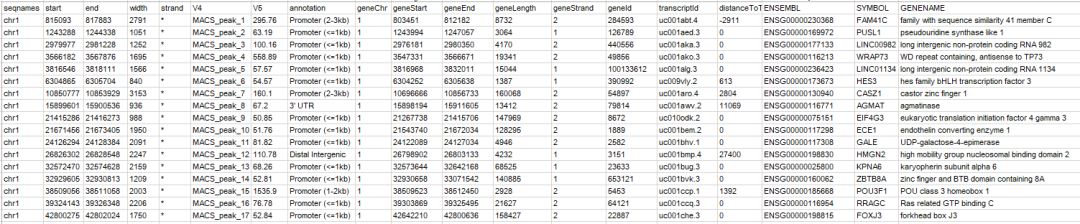
给出了关联的基因以及对应的基因组区域的类别,根据这个结果,可以提取关联基因进行下游的功能富集分析,比如提取geneid这一列,用clusterProfiler进行GO/KEGG等功能富集分析。
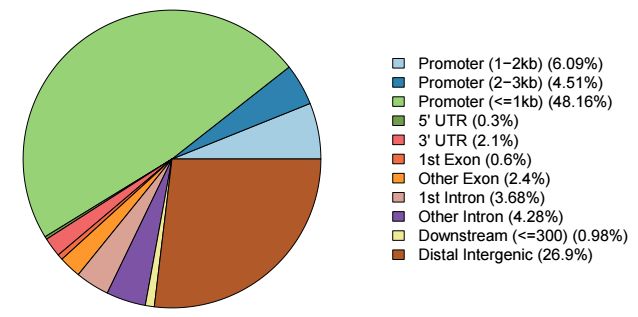
注释的结果还提供了多种可视化方式,其中饼图最为常见,用法如下
plotAnnoPie(peakAnno)
输出结果示意如下
以hg19为例,首先查询对应的GEO编号信息,用法如下
> hg19 <- getGEOInfo(genome="hg19", simplify=TRUE)
> head(hg19)
series_id gsm organism
111 GSE16256 GSM521889 Homo sapiens
112 GSE16256 GSM521887 Homo sapiens
113 GSE16256 GSM521883 Homo sapiens
114 GSE16256 GSM1010966 Homo sapiens
115 GSE16256 GSM896166 Homo sapiens
116 GSE16256 GSM910577 Homo sapiens由于列数太多,上述结果只展示了部分信息,对于每个bed文件,会列出对应的描述信息,方便筛选感兴趣的peak进行下载,可以根据GSM编号进行下载,用法如下
downloadGSMbedFiles("GSM521889", destDir="hg19")也可以根据下载一个基因组对应的所有peak文件,用法如下
downloadGEObedFiles(genome="hg19", destDir="hg19")
peak的overlap分析不仅可以探究生物学重复样本间的一致性,还可以进一步识别多种蛋白或者转录因子在调控网络中的作用,如果两个蛋白的chip结果overlap显著,很可能这两个蛋白构成了复合体,或者两种蛋白具有相互作用,这对于探究其调控机制有相当大的帮助。用法如下
enrichPeakOverlap(
queryPeak = peak_setA,
targetPeak = c(peak_setB, peak_setC),
TxDb = txdb,
pAdjustMethod = "BH",
nShuffle = 1000,
chainFile = NULL,
verbose = FALSE)依次将query的peak与target中的每一个peak文件进行overlap分析,计算出一个p值代表两个peak之间overlap的程度,p值越小,overlap的程度越高。
ChIPseeker除了peak基因注释的基本功能外,整合了GEO的下载功能与peak的overlap分析,可以方便的将自己的chip_seq数据与GEO的公共数据集进行比较分析。
感谢各位的阅读,以上就是“如何使用ChIPseeker进行peak注释”的内容了,经过本文的学习后,相信大家对如何使用ChIPseeker进行peak注释这一问题有了更深刻的体会,具体使用情况还需要大家实践验证。这里是亿速云,小编将为大家推送更多相关知识点的文章,欢迎关注!
免责声明:本站发布的内容(图片、视频和文字)以原创、转载和分享为主,文章观点不代表本网站立场,如果涉及侵权请联系站长邮箱:is@yisu.com进行举报,并提供相关证据,一经查实,将立刻删除涉嫌侵权内容。





